
Commençons pas la séance plénière.
La PKD foundation vient de donner 500 000$ pour la recherche en néphrologie via la fondation de l’ASN. Ce qui porte à 20 millions de $ les contribution de l’industrie ou des fondations privées pour la recherche, tu peux commencer à avoir une petite influence avec ce trésor de guerre.
Il y a ensuite la remise de l’Homer Smith award à Dontscho Kerjaschki. Il est viennois et anatomo-pathologiste. Il est connu pour ses travaux sur la néphrite de Heymann (modèle de GEM), il a découvert que l’antigène était la mégaline, malheureusement pour lui ce n’était pas le cas chez l’homme. Le podocyte humain n’exprime pas cette protéine. Il a participé à définir le role du complément dans la physiopathologie de la glomérulite extra-membraneuse et le role de la peroxydation des lipides dans la genèse des lésions de la membrane basale glomérulaire au cours de cette glomérulopathie.
Il s’est ensuite concentré sur les lymphatiques rénaux et ces vaisseaux de manière plus générale pour en devenir un spécialiste de renommée internationale. C’est en travaillant dans ce domaine qu’il a fait un retour vers ses premières amours néphrologiques. Son groupe a créé une souris transgénique surexprimant un miRNA le 193a pour étudier son rôle dans la lymphogenèse. Ces souris présentent des lésions du podocytes avec apparition d’une hyalinose segmentaire et focale. Le mIR-193a contrôle l’expression de tous les gènes importants pour l’architecture podocytaire. Sa surexpression entraine une sous expression expliquant le développement de la maladie rénale. Ce miR est augmenté chez certains patients avec une HSF. Chez la souris le blocage de l’expression du miR par un LNA permet de prévenir la progression. Le résultat le plus excitant est, normalement si les podocytes n’expriment pas le miR-193a, les cellules pariétales de la capsule de Bowman oui. Quand on diminue son expression elles prennent un phénotype podocyte-like. Lors d’une glomérulopathie expérimentale faisant des croissants, il y a une surexpression du miR. Son inhibition empêche la formation des croissants. En écoutant le topo je me demandais si on pouvait le détecter dans le sang ou les urines le miR-193a pour en faire un biomarqueur de certaines maladies glomérulaires. Un très intéressant talk qui confirme l’importance des miR et de la serendipité en biologie.
La présentation état de l’art a été faite pas Helen H. Hobbs. Les anticholestérols doivent sauter ce paragraphe sous peine d’éruption urticarienne voir de choc anaphylactique. 50% de la causalité de la pathologie coronarienne est génétique. L’orateur a construit sa carrière sur le cholestérol. Elle rappelle ce graphique qui montre l’importance du cholestérol dans la genèse de la pathologie coronarienne du moins à Framingham. 90% des patients avec un CT ont présenté une coronaropathie alors qu’ils ne représentent que 3% de la cohorte totale. Le chevauchement des courbes expliquent pourquoi pour voir un effet dans les essais interventionnels il faut inclure quelques milliers de patients. L’hyperCT est responsable de coronaropathies, ceux qui en doute devrait de temps en temps voir des patients avec des hyperCT familiales graves. Le cholestérol n’est pas le seul intervenant mais il joue un rôle indéniable.
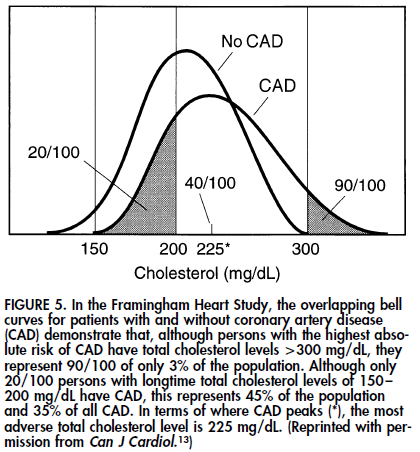
L’originalité de l’approche de l’auteur est de s’être concentrés sur les outliers à la recherche d’une hérédité mendélienne dans les anomalies du LDL-C. Coté trop, il y a des pathologies dominantes avec un défaut d’expression du LDLD-R et des mutations dans ApoB, des pathologies récessives, des mutations dans ARH, nécessaire pour l’endocytose du LDL-R, et la sitsterolémie : ABCG5 et ABCG8 servent à faire sortir le CT dans la bile. L’augmentation du LDL dans ces maladies génétiques entrainent une coronaropathie quelque soit le mécanisme.
Est-ce que le LDL est indispensable pour faire une coronaropathie ? Elle a construit une étude de population où elle va chercher des patients avec un LDL bas. C’est la Dallas Heart Study, 3557 patients avec 51% d’afro-américians. Le but: séquencer les extrêmes, HDL bas (mutation dans ABCA1) et LDL haut (mutations dans NPC1L1). Après la création de la cohorte une nouveau gène muté, PCSK9, donne une hyperCT dominante. Une protéine secrétée dont la surexpression entraine une diminution du LDL-R et une augmentation du LDL. Il s’agit de mutations gain de fonction. Ce qu’a montré le Dr Hobbs c’est que des mutations tronquantes entrainant une diminution de la sécrétion de PCSK9 diminuent le LDL-C. 2 % des afro-américains portent des stop qui diminuent de 40% le LDL circulant. 3% des caucasiens portent une mutation, R46L, diminution de 21% de LDL. L’analyse de la cohorte ARIC montrent que chez les AA, on observe une diminution de 28% du LDL-C et une diminution de 88% du risque d’événement CV, chez les caucasiens, le LDL diminue de 15% et diminution de 46% du risque CV. Ces résultats ont été répliqués dans trois cohortes indépendantes. Les mutants PCSK9 font mieux en terme de réduction du risque que les statines, c’est l’effet temps. Le génotype est une intégration du LDL par le temps. Elle parle de « LDL iceberg effect ». Avoir moins de 150 mg de cholesterol entraine un risque d’IDM de 0,1%. Les statines ont un effet non optimal sur le LDL-C car l’augmentation du LDL-R induite par l’augmentation de SREBP-2 est contrebalancée par une augmentation de PCSK9 qui diminue l’expression du LDL-R. Ces travaux ont ouvert la porte à l’utilisation des ac anti-PCSK9, il y a actuellement 5 anticorps en développement par tous les acteurs majeurs de la pharmacie, avec des résultats intéressants sur la diminution du LDL-C. Je vous rappelle pour aller plus loin sur le sujet, cette note. En passant le prix des antiPCSK9 devrait être de 14000$ par an et par patient, je vous conseille la lecture de cet édito du NYT.
Je suis allé voir la session biologie de système.
La première présentation portait sur la génomique en transplantation avec la GoCAR study. En pratique, ce groupe a montré qu’il pouvait prédire aussi bien la progression de la fibrose que le risque de rejet avec une approche intégrative de transcriptomique. Il arrive avec 13 gènes à prédire le risque de développement de la fibrose et avec 11 gènes à prédire le risque de rejet infracliniques. Ces approches permettront probablement de mieux prendre en charge les tranplantés sans avoir recours à des biopsies. Un jour nous ferons de la transcriptomique en routine chez les transplantés rénaux et nous modifierons le traitement sans biopsie.
La présentation suivante par Matthias Kretzler pour le groupe Neptune nous présente la médecine de précision au service du syndrome néphrotique. Ils ont inclus dans 23 centres aux USA, 460 patients (30% de moins de 18 ans) 268 HSF/LGM, 74 GEM et d’autres néphropathies. Le but est d’identifier de nouveaux groupes de patients, en croisant les données de la biopsie, sanguines, urinaires et les données cliniques. Il y a un suivi de 5 ans avec prélèvements bio tous les 6 mois pour de la génomique, protéomique etc. Il passe d’une analyse en une dimension en une analyse en 3D. On le faisait déjà mais les omics permettent d’amplifier la démarche. Il donne comme exemple l’impact du génotype d’APOL1 sur le SN qui vient d’être publié dans JASN en 2015.
Leur but est d’identifier des biomarqueurs pour mieux stratifier les patients ce qui permettra de faire des essais uniquement sur ceux à risque de progression pour diminuer le volume des essais et n’inclure que des patients qui pourraient bénéficier de la molécule. Il donne l’exemple non publie de l’EGF, une approche de transcriptomique sur biopsie a permit de montrer que sa surexpression dans le rein est associé à la progression de l’insuffisance rénale. Ils ont ensuite pu montré que le taux dans les urines reflète le tissu. La concentration d’EGF urinaire est prédictif de la fibrose rénale et du risque de dégradation du DFG.
Il finit avec la dissection moléculaire du groupe HSF/LGM en utilisant la transcriptomique. Ils ont identifié trois clusters de gènes. L’un d’eux permet d’identifier des HSF de mauvais pronostic. Au cœur du réseau, il y a le TNF. Ils ont confirmé ces résultats dans une cohorte indépendante. Ce qui veut dire qu’on pourrait justifier de recommencer un essai avec l’Adalimumab en ne le donnant qu’aux patients du cluster 3. Il montre qu’il peuvent avoir les mêmes résultats qu’avec la biopsie rénale juste en regardant les urines, qui devient une biopsie liquide grâce à la technique Luminex.
Ces travaux ont été possible en adaptant l’approche Transmart dans les maladies rénales.
K Sharma vient nous parler du métabolome dans les maladies rénales. Ce fut un excellent talk. Il a une manière très amusante de présenter le boulot. L’impression générale est on met n’importe quel liquide biologique dans le mass spect et on trouvera toujours quelques choses. Il explique très bien les limites de l’approche, techniques différentes, pas de haut débit, pas d’automatisation, pas de bioinformatique, la spécificité, la linéarité, la reproductibilité et l’absence bien souvent de standards.
Il n’empêche qu’il nous montre des résultats magnifiques publiés dans JASN. Ils ont identifié 12 métabolites prédisant la dégradation de la fonction rénale. Par une approche de système (utilisant cytoscape) ils trouvent que le lien entre les métabolites est la mitochondrie. La néphropathie diabétique s’accompagne d’une perte de mitochondries liée à un défaut d’expression de PGC1α.
Ils ont confirmés et amplifiés ces données dans des néphropathies non diabétiques. La fonction mitochondriale est encore altérée par un déficit en PGC1. Dans un autre modèle, il y a une augmentation du ratio ATP/amp conduisant à une augmentation de l’activité glycolytique au dépend de la respiration mitochondriale. Si on active AMPK on diminue l’albuminurie.
De façon intéressante, quand on surexprime NOX4 dans les podocytes d’une souris elle fait une néphropathie diabétique. Les inhibiteurs de NOX4 améliore la néphropathie diabétique. Le fumarate diminue ainsi de même que le TGFb. De plus MDM2 serait au cœur de la régulation des processus précédemment décrit jouant un rôle de de chef d’orchestre du métabolisme rénal. Ces phénomènes me rappellent un papier lu récemment. Il faut que je creuse cette piste. Le sentiment plus général est que la transition glycolyse/phosphorylation oxydative est au cœur de la pathologie du podocyte. Si on ajoute les données observées dans la polykystose rénale, les maladies rénales sont très métaboliques…
Nous finissons avec une présentation sur la protéomique par Jon B. Klein. Le message est identifier des protéines c’est compliqué. L’ensemble de la présentation ne sera que de nous dire à quel point la technologie n’est pas au point pour la routine. Il faut savoir que 80% des protéines des urines sont des protéines du plasma. Il conseille l’immunodéplétion. Il souligne l’intérêt de travailler sur les exosomes. Devant un syndrome néphrotique la présence d’exosomes portant LMP2 permet de différencier GEM d’HSF. Le préanalytique est capital. Enfin il nous cite une technique d’avenir la SWATH-MS. Un topo trop technique.
Après la pause déjeuner, une session hémostase, je me suis régalé. Je ferai une note à part sur ce sujet car il me semble très important. Je me demande si je n’ai pas un petit conflit d’intérêt en disant ça.
Pour finir une petite session d’abstract en présentation orale sur l’hypertension.
Première présentation, une étudiante en médecine qui vient présenter des données sur la variabilité de la tension artérielle systolique et ses effets sur la fonction rénale. Une étude de cohorte de 2 865 157 patients avec un DFGe à deux reprises et 8 mesures de PA. Après un ajustement sur une multitude de cos variables, la variabilité de la TAS est prédictive de la mortalité, de la mortalité CV et de l’insuffisance rénale chronique terminale. Je ne sais pas ce qu’on en fera mais si vous voulez publier en néphro travaillez sur la variabilité tensionnelle, ça à la cote.
Après la très jeune étudiante en médecine voici un très jeune chercheur sur la polykystose rénale autosomique dominante, Vincente Torres. Il s’agit d’une sous étude de HALT PKD étudiant l’impact d’un contrôle strict de la pression artérielle sur l’hypertrophie ventriculaire gauche. Les sujets ont un DFG normal et de façon très étonnante, en IRM cardiaque, une fréquence de l’HVG très faible. Sur les 543 patients moins de 1% ont une HVG, le suivi est de 60 mois. La masse ventriculaire gauche diminue avec le traitement intensif de la pression artérielle. Je ne sais pas ce que cela veut dire alors qu’on a une masse VG normale de la voir diminuer encore. Si un cardiologue lit ses mots j’aimerai avoir son avis.
On continue avec les jeunes, Carmine Zoccalli vient nous parler de l’interaction système sympathique, ADMA, SDMA. En pratique 14 patients sans insuffisance rénale avec dénervation rénale suivi 6 mois. La dénervation diminue l’activité sympathique qui fait diminuer les taux circulant d’ADMA et SDMA. Une présentation didactique qui n’en finit pas par un vieux renard, j’aime bien, même si l’impact clinique ne me parait pas évident.
Avant la présentation de SPRINT, encore un peu de contrôle TA strict contre habituelle dans la cohorte SPS3. 3020 personnes avec des lacunes dans le cerveau l’objectif est de comparer 130-149 à <130 de systolique. Il n’y a pas eu d’impact sur la récurrence des AVC. Est-ce que ça un effet sur le rein? C’est un post hoc non spécifié. Il y 2610 patient avec au moins 2 créat. 3 ans de suivi, 16% DFG<60. A un DFGe diminue de 2 ml/mn de plus dans le groupe intense après la première année plus grand-chose. 24 % contre 19% ont diminution rapide dans groupe intense comparé à l’habituelle et de façon statistiquement significatif. Je ne trouve pas les résultats bien passionnant si ce n’est dire que faire baisser la pression artérielle fait diminuer le DFG, niveau physiologie rénale de 3é année de médecine.
L’étude qui sera bien publiée maintenant car techniquement c’est un tour de force. Travail norvégien. 1299 patients ont eu à 5,6 ans d’intervalle la mesure du DFG par un iohexol. Le DFG de départ est normal: 102 ml/mn. Il y aura une diminution de 0.6 à 1.2 ml/mn/an du DFG en fonction de la TA. Il trouve après des gesticulations statistiques que ceux qui sont normotendus dégradent plus vite que les autres. C’est du vent, la diminution de DFG est tellement ridicule que je ne crois pas que nous soyons dans le domaine du pathologique. Une excitation pour vraiment pas grand chose, que d’argent dépensé inutilement.
Le meilleur abstract de la session pour finir, Est-ce que l’allopurinol fait diminuer la TA? Quand fait monter l’acide urique chez un rongeur, on observe une augmentation de la pression artérielle. Une méta analyse chez l’homme montre un lien AU et HTA. Ce travail, la MODERATE study, s’intéresse à des obèses, hyperuricémique, avec des TA <140/90. On leur donne soit du probenecid, soit de l’ allopurinol soit un placebo. Il y a 50 patients par groupe. Après 8 semaines, il est observé une diminution de l’acide urique mais strictement aucun effet sur la TA, sur le RAS et sur la fonction endothelial (FMD). Ce travail montre que l’acide urique n’est pas causal mais est juste associé. L’orateur s’est vu déboulé des défenseurs de l’acide urique, les débats ont été chauds, mais il s’en est très bien sorti. Je félicite Mr McMullan d’avoir tenu.
Ce fut ma dernière écoute de la journée.

Hello,
« par contre 90% des personnes avec plus de 3 g de cholesterol ont eu une coronaropathie. »
Je ne comprends pas. Pour moi il y a une discordance entre le graphique et son explication.
Merci de m’éclairer
Bonjour. Merci de vos compte-rendus.
Tout cela est très intéressant, mais je ne suis pas sûr malheureusement que cela va changer à grand-chose aux patients. Hormis l’indispensable création de réseaux et cohortes de glomérulopathies pour avancer sur ce terrain particulier, sujet pour lequel nous paraissons en retard en France.
Ce qui me trouble sinon est depuis des années la mise en avant des biomarqueurs, des marqueurs génétiques, -omiques et autres censés améliorer la prise en charge de néphropathies, la néphropathie diabétique tiens par exemple, on dirait parfois de la pensée magique. Depuis 10 ans que je suis dans le milieu néphrologique les jolies molécules et techniques défilent dans les publications et à la fin c’est des IEC, ouah… Bon la critique pour un non-chercheur est facile mais on a l’impression qu’on se gargarise de nouveauté technique faute de nouveauté thérapeutique… .
8 semaines est-ce assez pour juger définitivement des liens urate-TA? Question complexe que celle-là.
Enfin comme le dit nfkb le graphique est alambiqué et pas vraiment aidé par la légende. On aurait envie de dire 100% en se fiant à la courbe. Quant à « the most adverse CT level is 225 mg/dl », qui mélange fréquence de distribution et risque c’est vilain. Dommage d’embrouiller un message qui a déjà du mal à passer.
Bonne journée
Je pense que parmi 100 biomarqueurs moins d’une dizaine seront utilisables en clinique et probablement un ou deux en pratique. La CRP a commencé comme ça, la créatinine et je ne parle pas du BNP ou de la troponine. Pour la néphropathie diabétique, ça tombe bien, un game changer est apparu avec des résultats cliniques stupéfiants. J’y reviendrai. Pour HTA et urate la réponse est 8 semaines suffisent à mon avis largement en plus.
Mon avis de cardiologue sur la question de la masse myocardique
En échocardiographie, on classe la géométrie ventriculaire en normale, hypertrophie concentrique, hypertrophie excentrique et remodelage concentrique. La différence se fait selon l’existence d’une augmentation ou non de la masse ventriculaire indexée, et sur une modification de l’index d’épaisseur relative (index h/r, qui rapporte l’épaisseur de la paroi postérieure au diamètre ventriculaire). Le remodelage concentrique est défini par une masse normale mais avec une épaisseur relative augmentée. A ma connaissance, cette donnée n’est pas couramment utilisée en IRM. Il faut aussi savoir qu’il existe des hypertrophie septale localisée sans augmentation de la masse ventriculaire ni de l’index d’épaisseur relative.
Dans l’affaire qui nous intéresse, en l’absence d’hypertrophie initiale, la baisse de la masse n’a aucune incidence ou importance particulière (l’ampleur de cette baisse a-t-elle au passage une signification clinique ?). Éventuellement, elle pourrait s’accompagner d’un remodelage inverse en cas de remodelage concentrique initial, comme on le voit dans certaines cardiopathies post-hypertensives débutantes, mais il n’est pas mentionné d’index h/r dans la publication initiale.
Donc, le seul intérêt est de montrer qu’il n’y a pas d’apparition d’hypertrophie sous traitement (ce qui est rassurant…)
merci
Ping : HOPE-3 ou une statine n’est qu’une statine | PerrUche en Automne